Gviz, Visualize genomic data. Let’s dive into it to explore useful functions.
Introduction to Gviz package
- Provide a structured visualization framework to plot any type of data along genomic coordinates.
- Integrate publicly available genomic annotation data from sources like UCSC or ENSEMBL.
Plot annotation track
|
|
output
|
|
output

Add genome axis track
|
|
output

Add chromosome ideogram
|
|
output
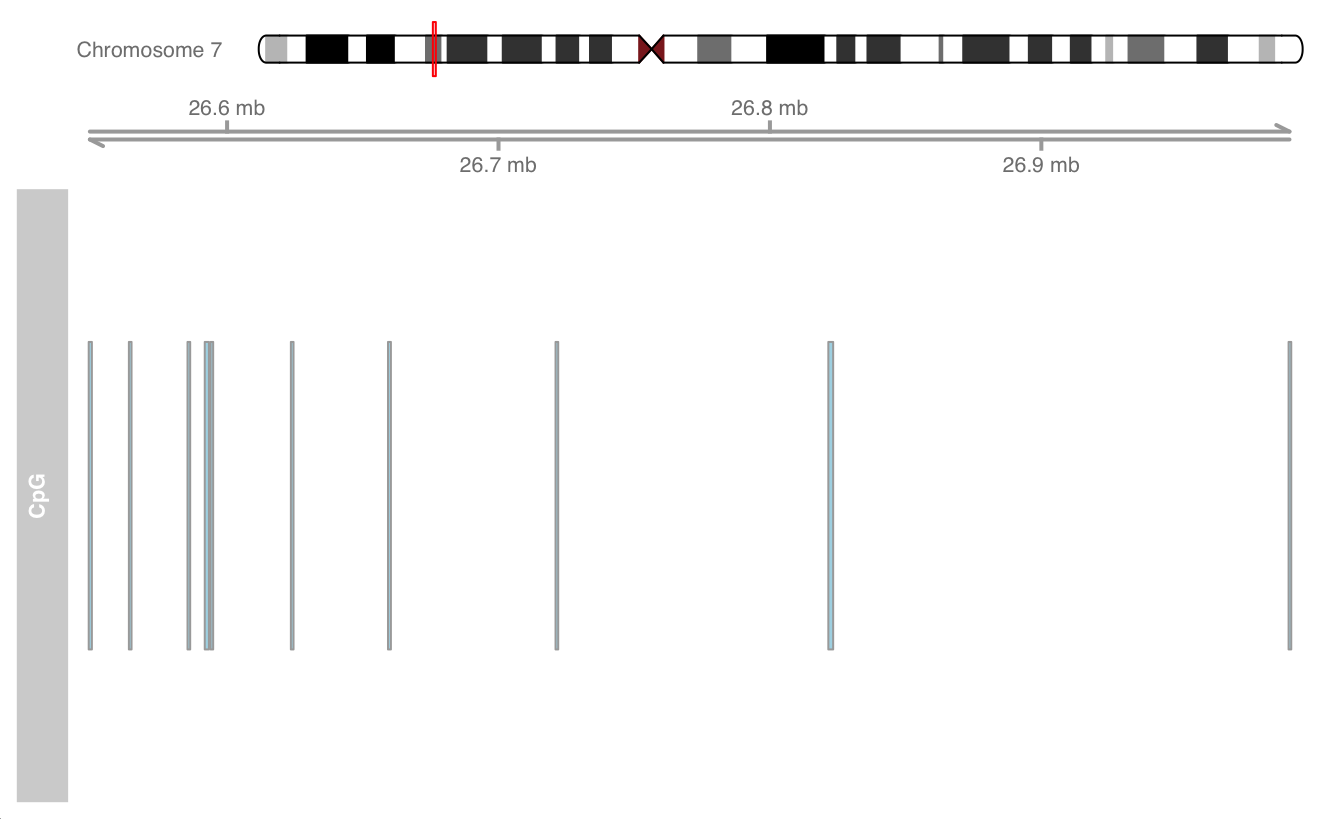
Add gene model
|
|
output
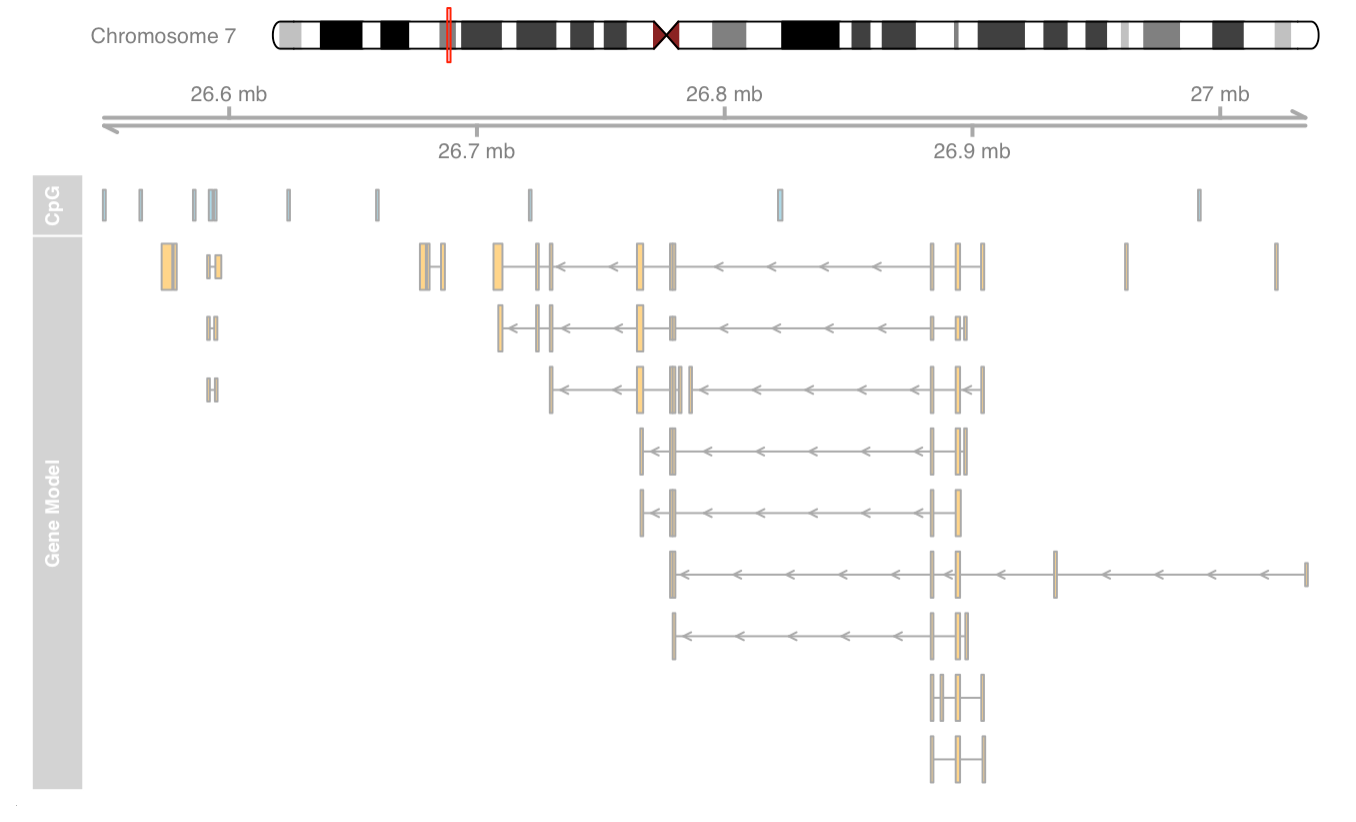
Zoom the plot
|
|
output
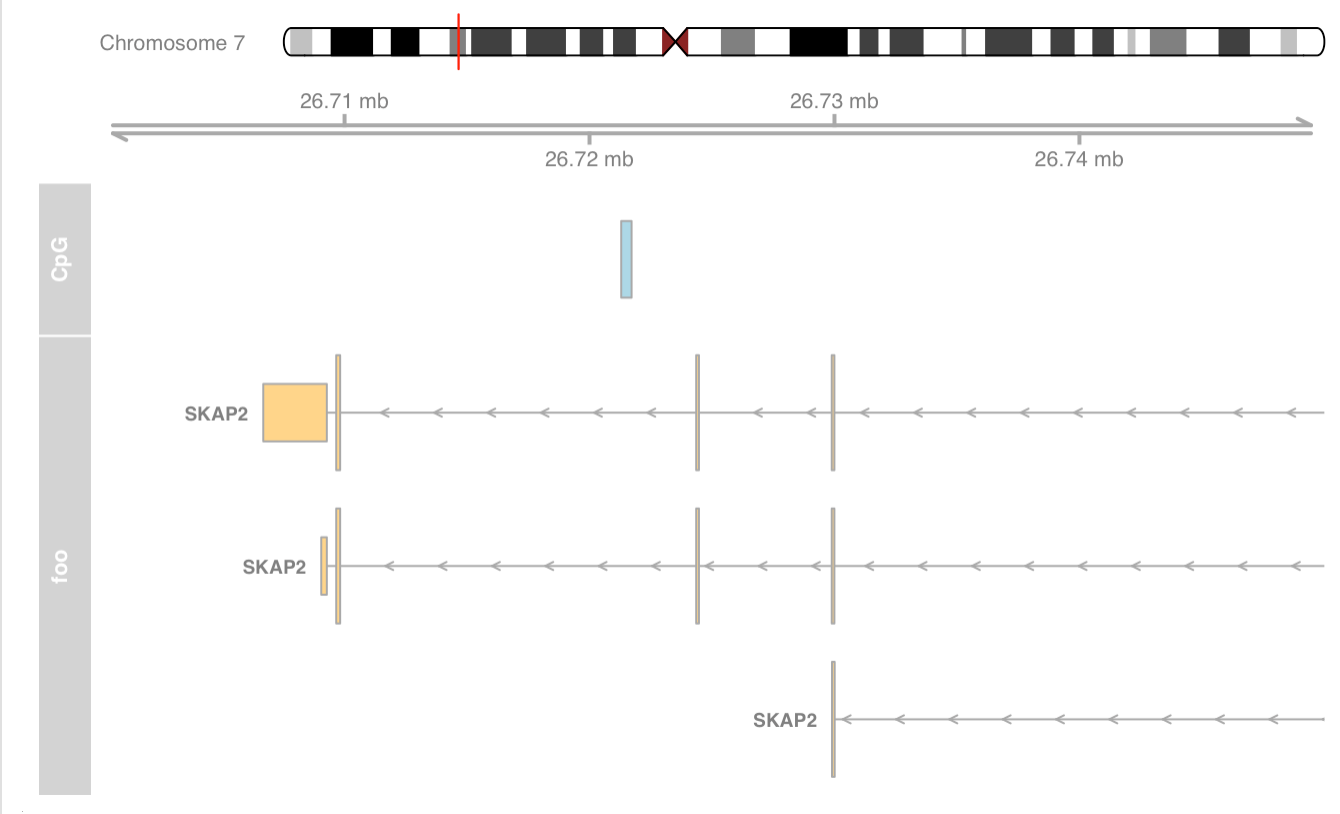
|
|
output

|
|
output
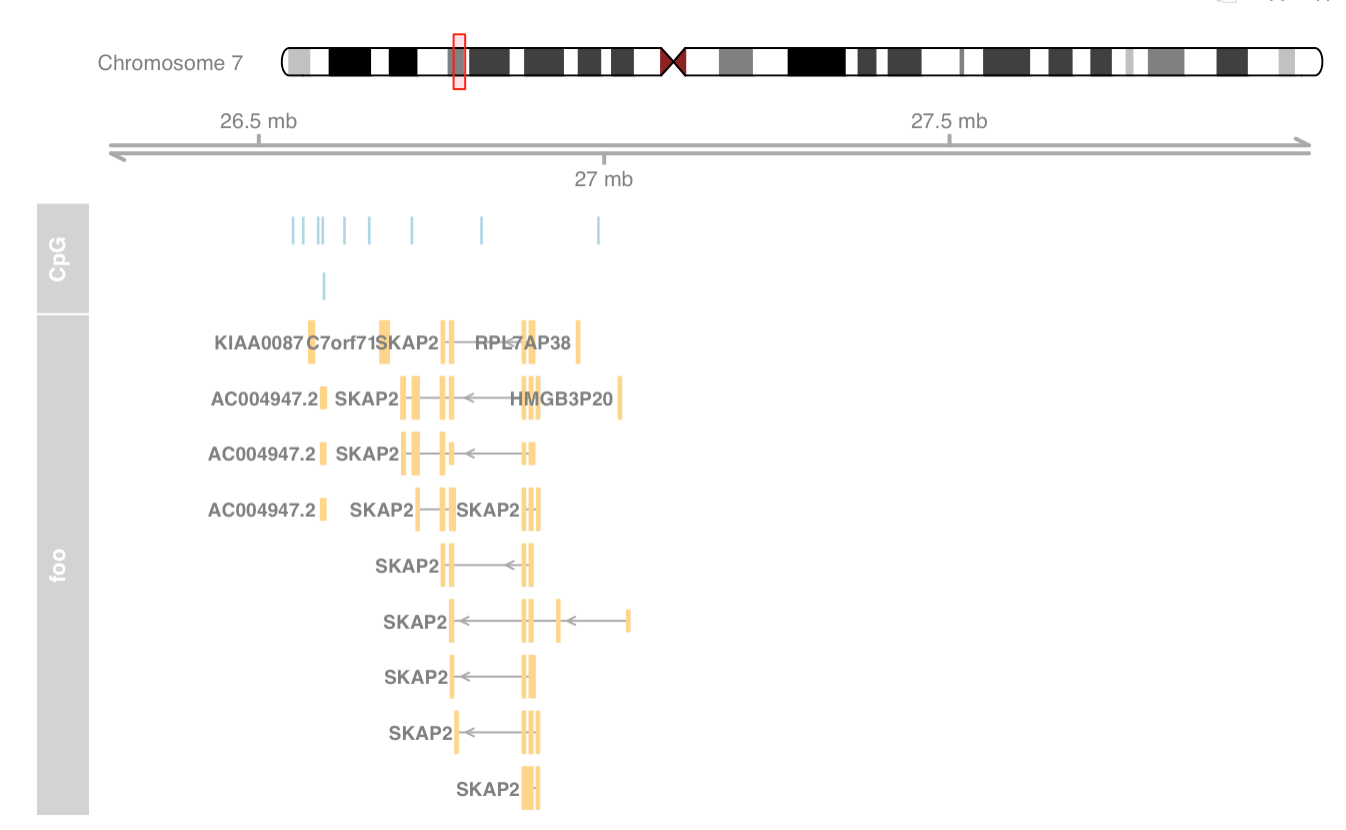
Add sequence track and zoom to view sequence
|
|
output
Add data track
|
|
output
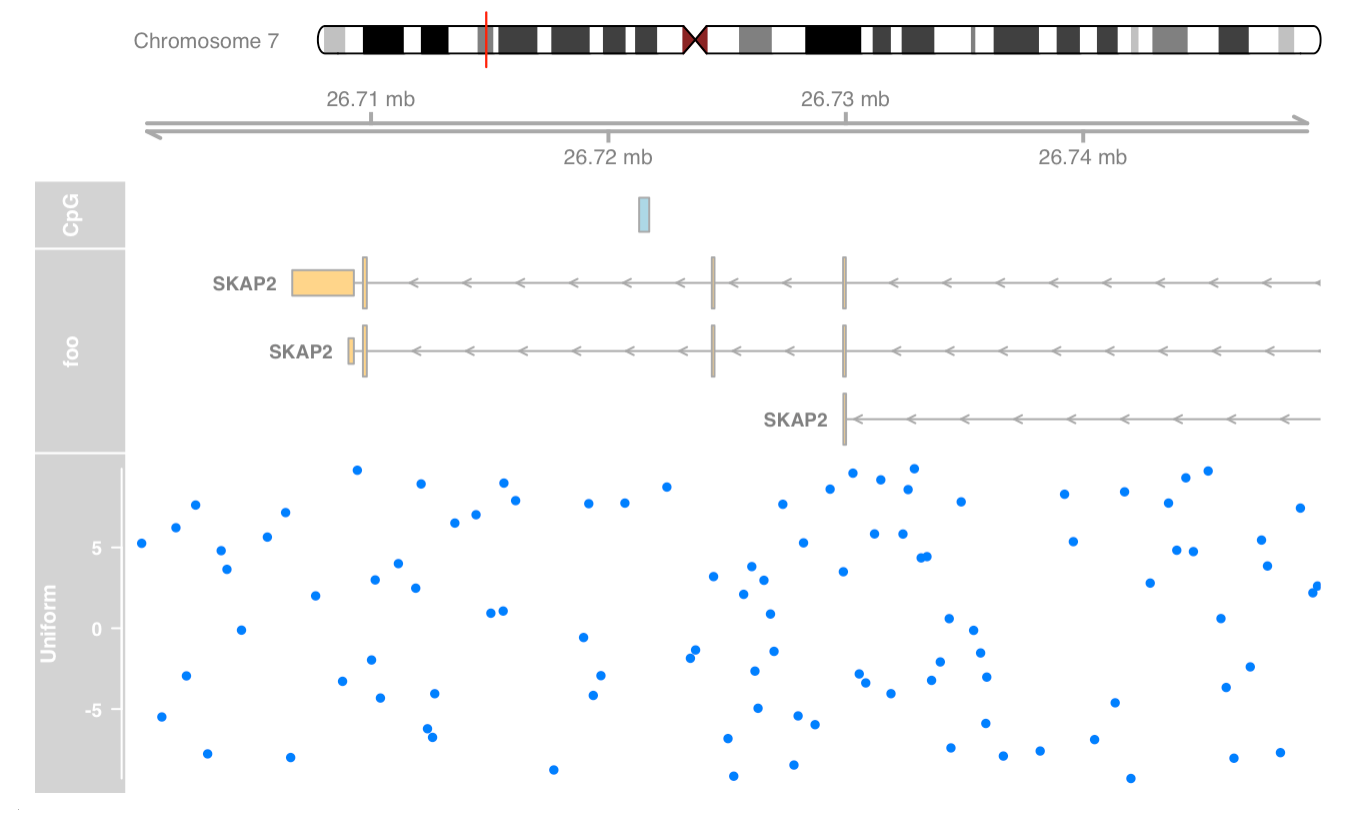
|
|
output
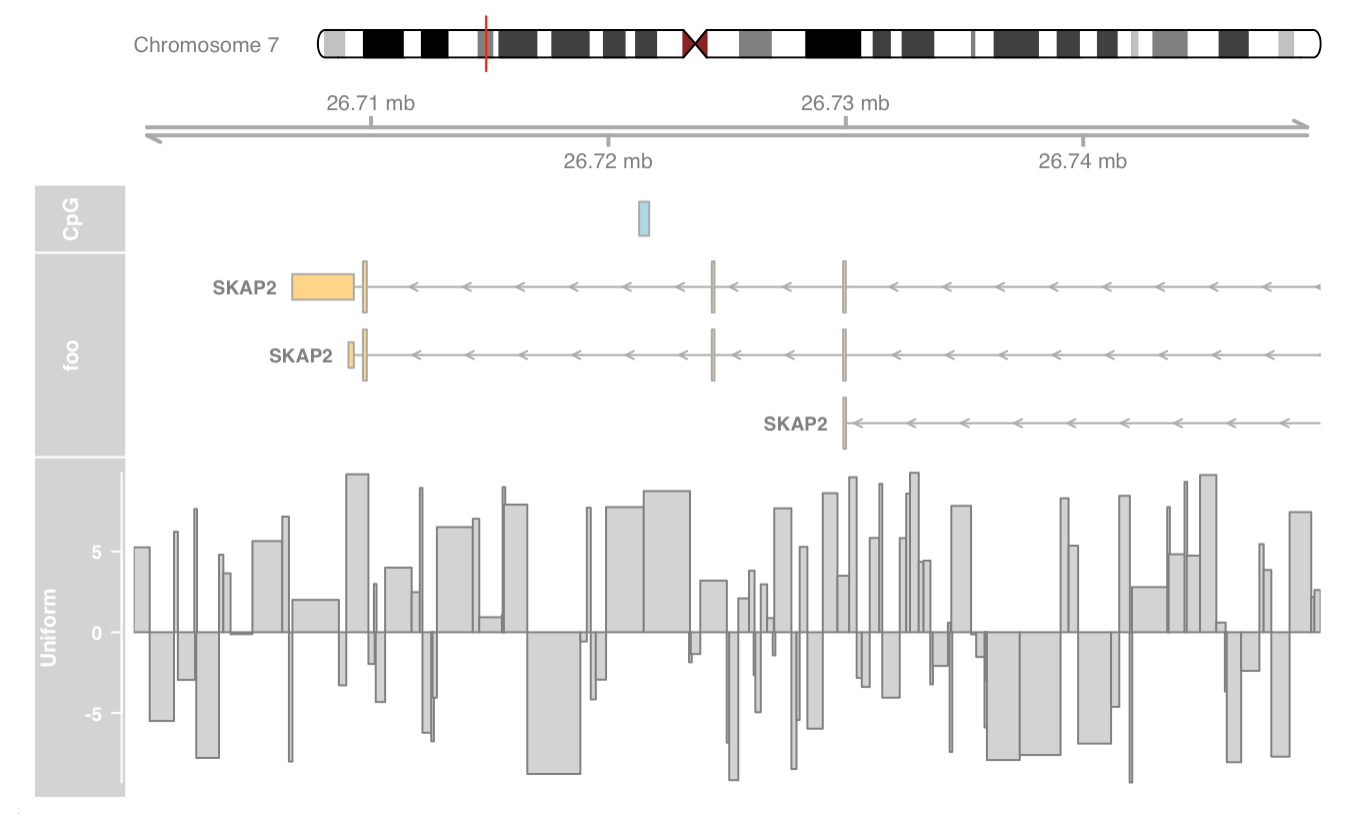
Plotting parameters
|
|
output
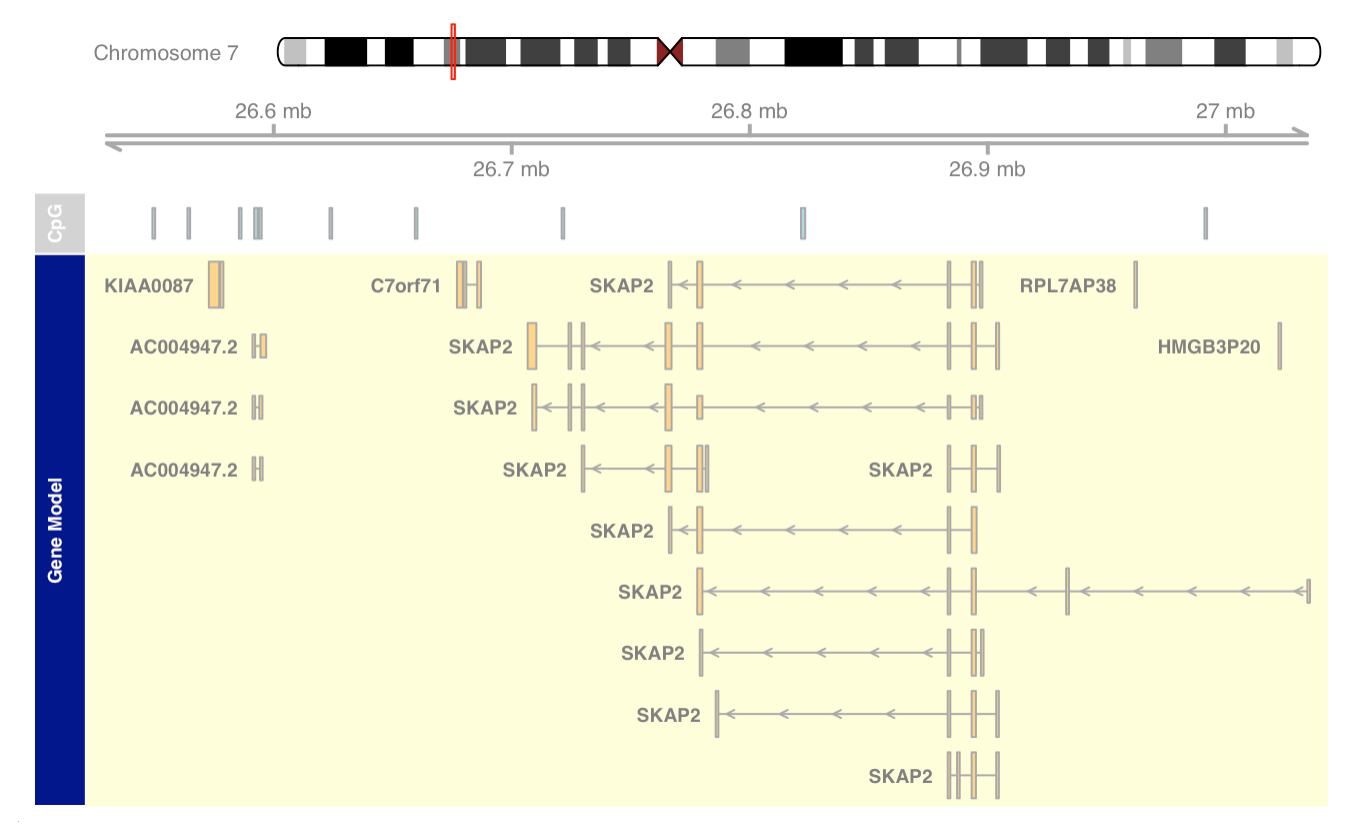
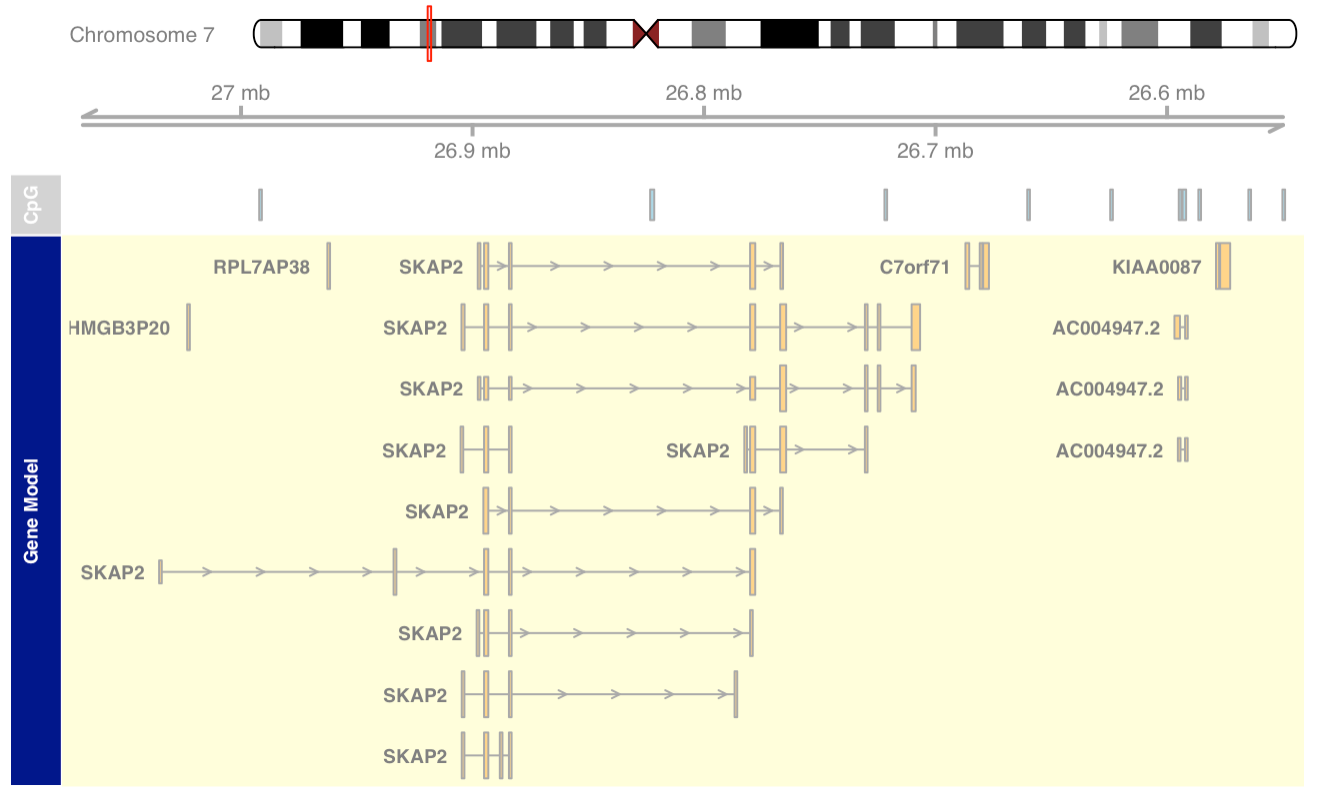
Plotting direction
|
|
Track classes
|
|
output

IdeogramTrack
|
|
output
|
|
output
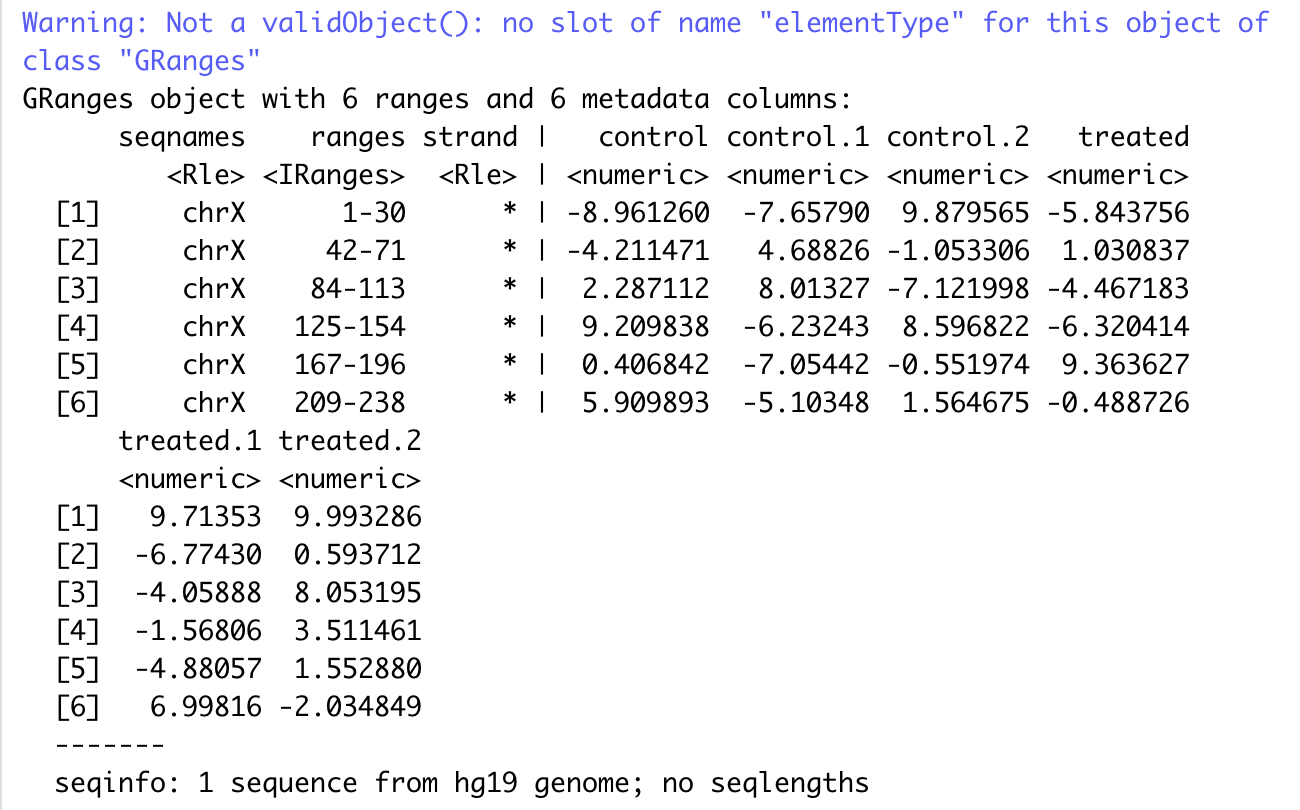
DataTrack
|
|
output
|
|
output
|
|
output
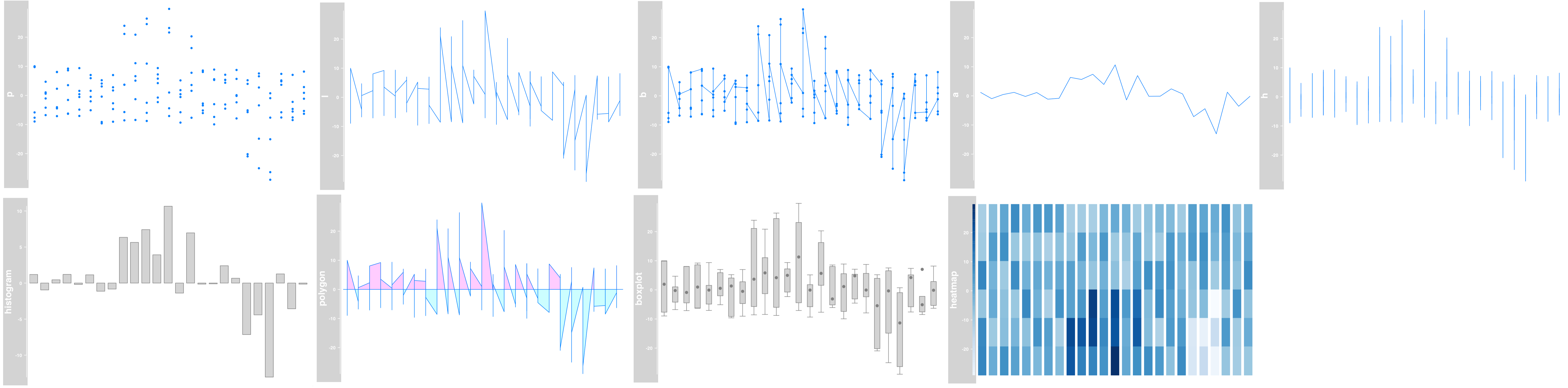
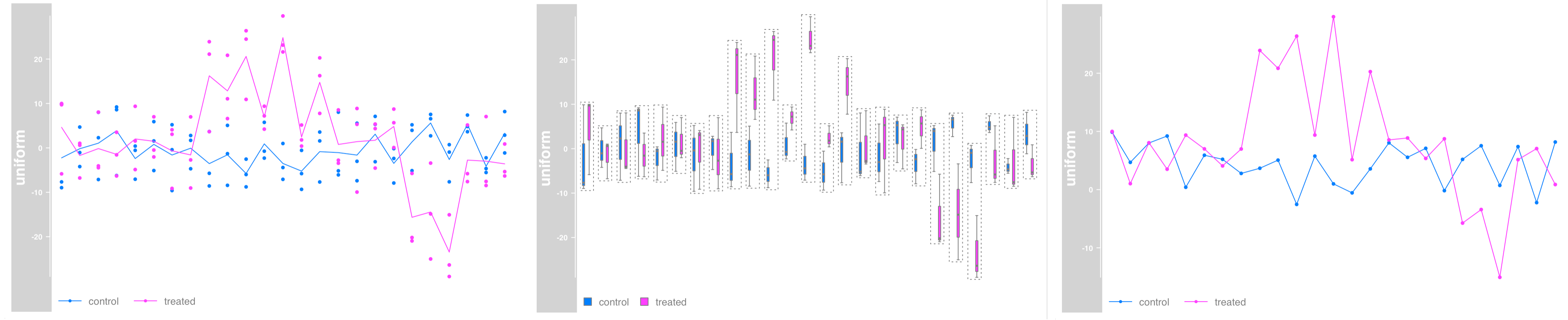
Example of DataTrack plots
|
|
output
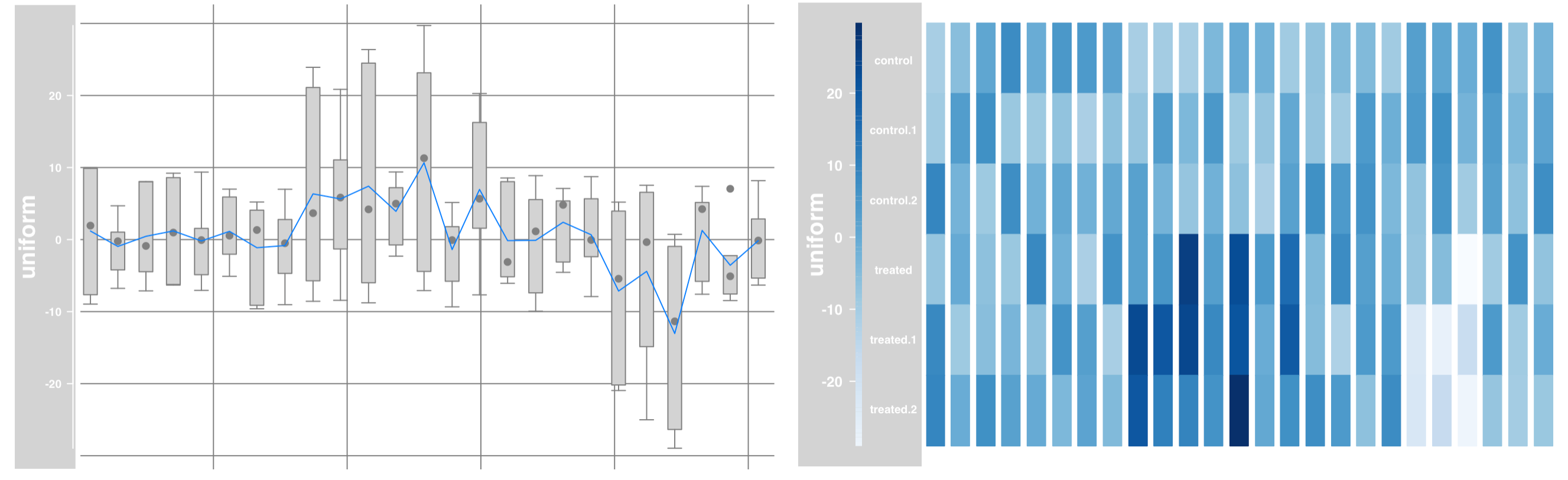
Data grouping
|
|
output
Building DataTrack objects from files
|
|
output
AnnotationTrack
|
|
output
Add id information
|
|
output
change fontcolor
|
|
output
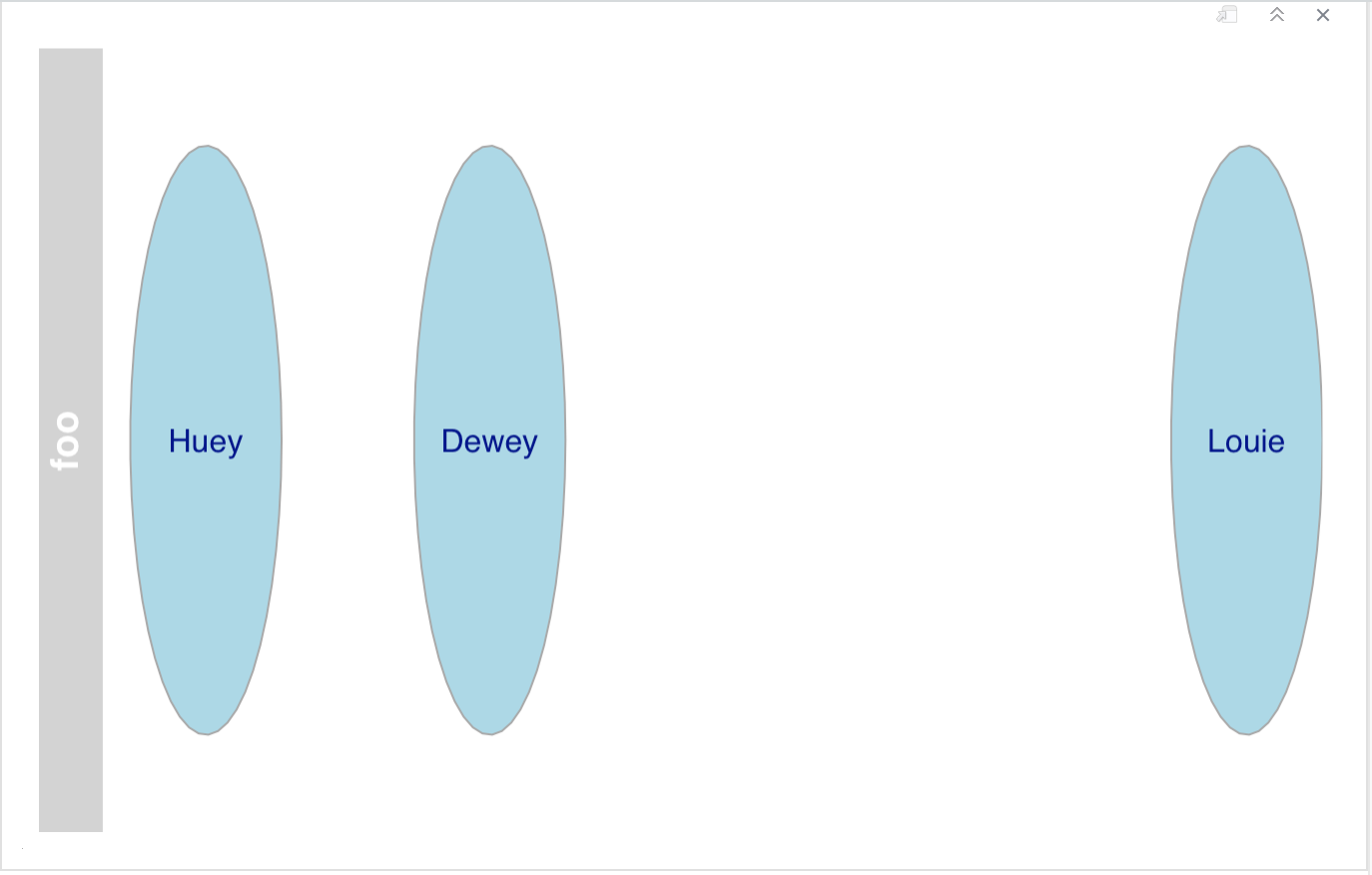
|
|
output

plot both the DataTrack representation as well as the AnnotationTrack representation of the bam file together
|
|
output
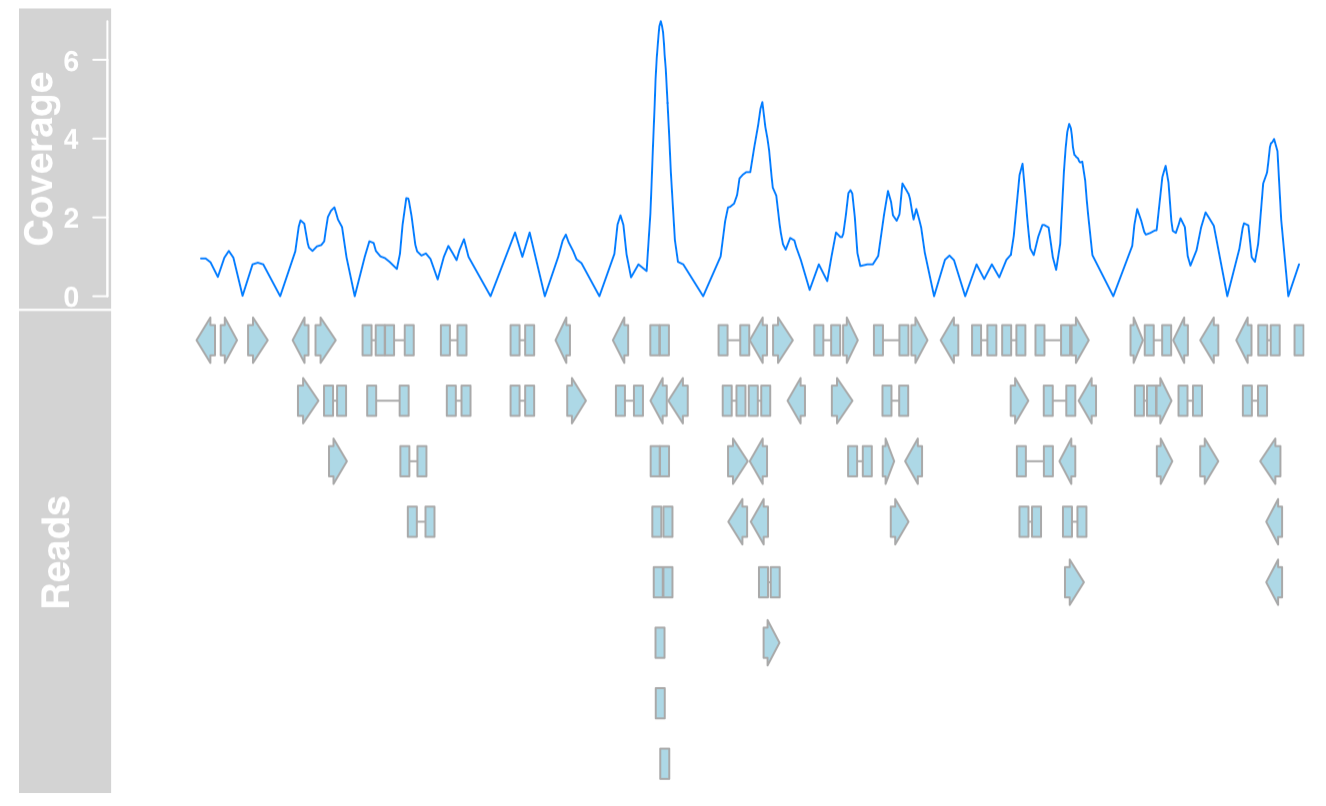
GeneRegionTrack
|
|
Building GeneRegionTrack objects from TranscriptDbs
|
|
BiomartGeneRegionTrack
|
|
output

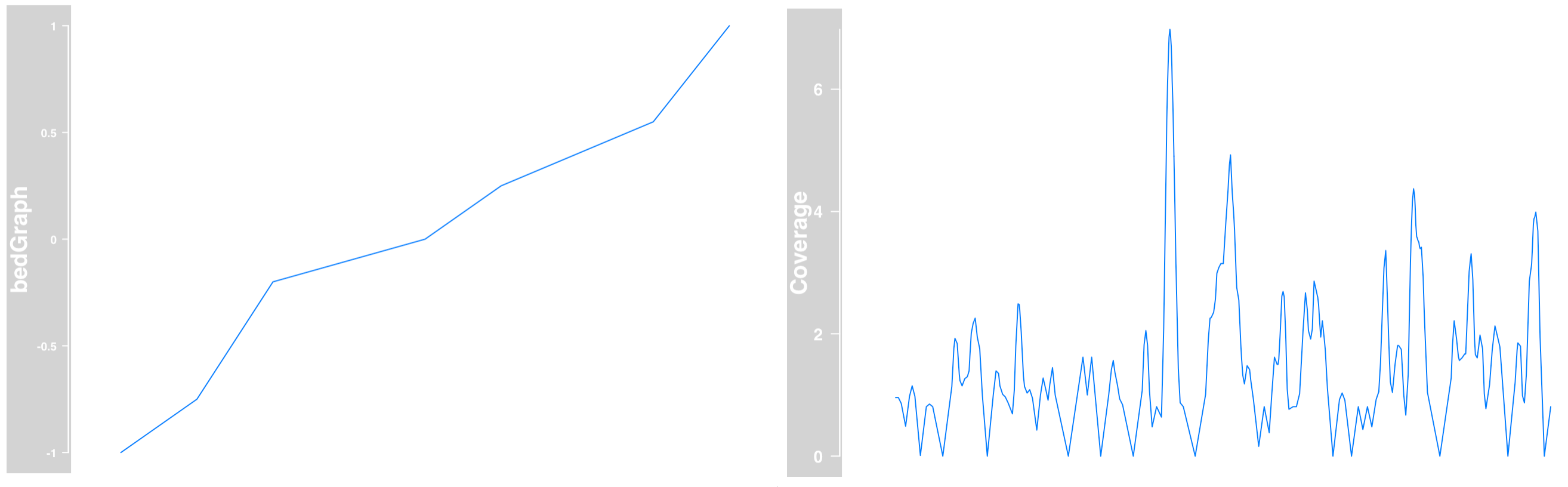
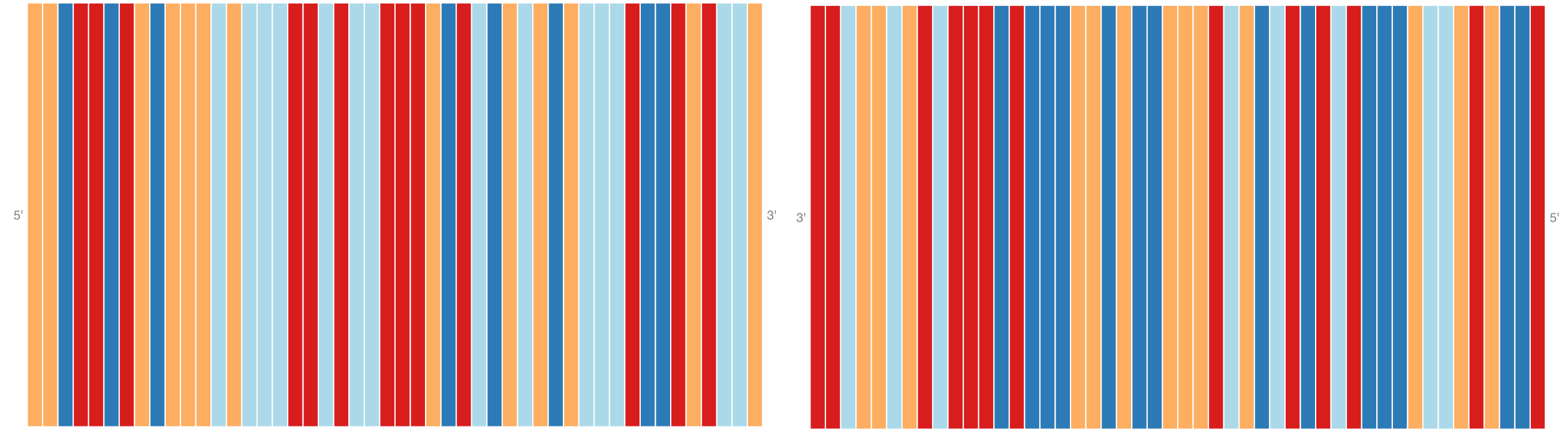
Sequence Track
|
|
output
AlignmentsTrack
RNAseq experiment
|
|
output
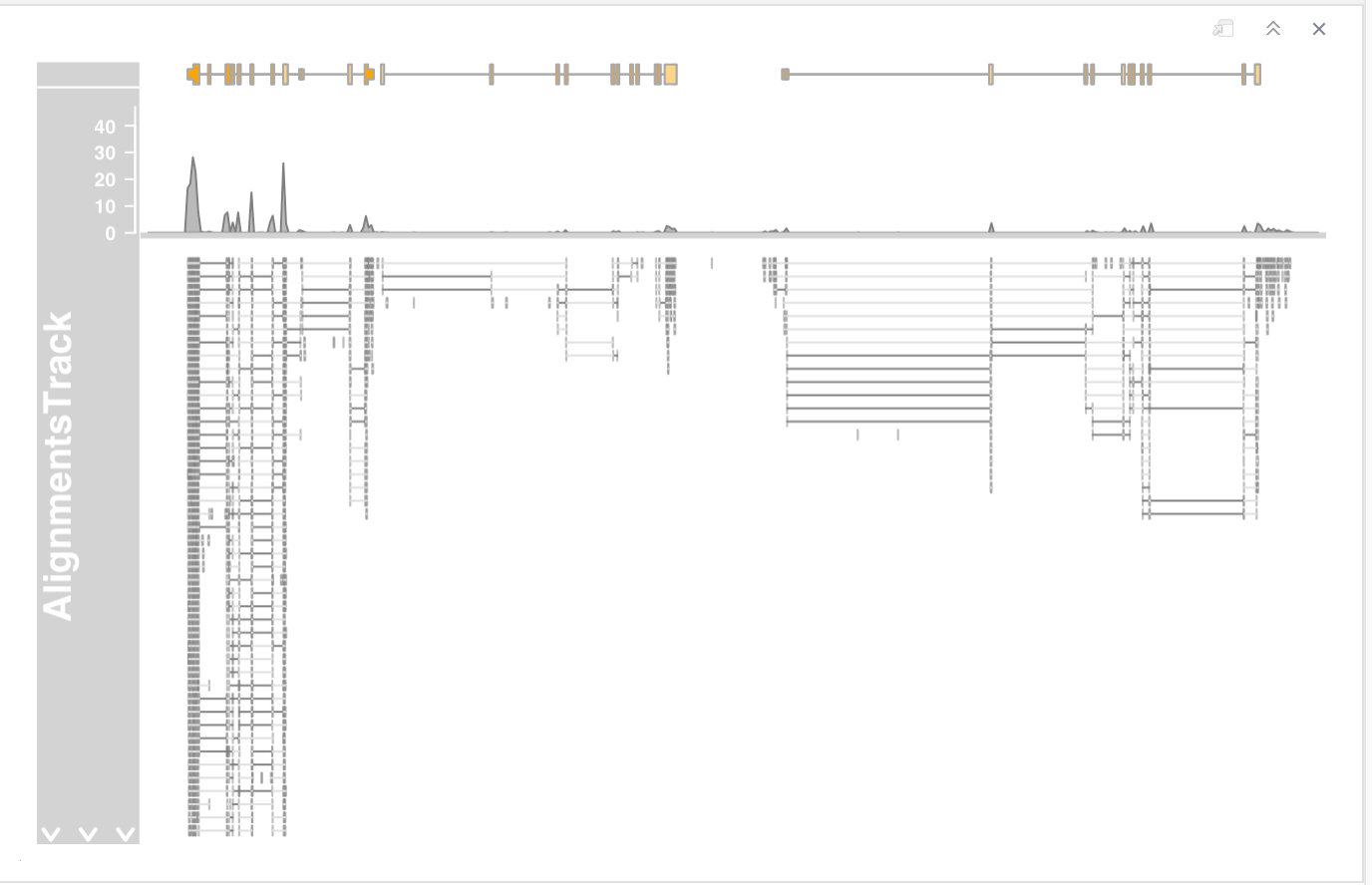
- To reduce the size of the coverage section by setting the coverageHeight or the minCoverageHeight parameters.
|
|
output
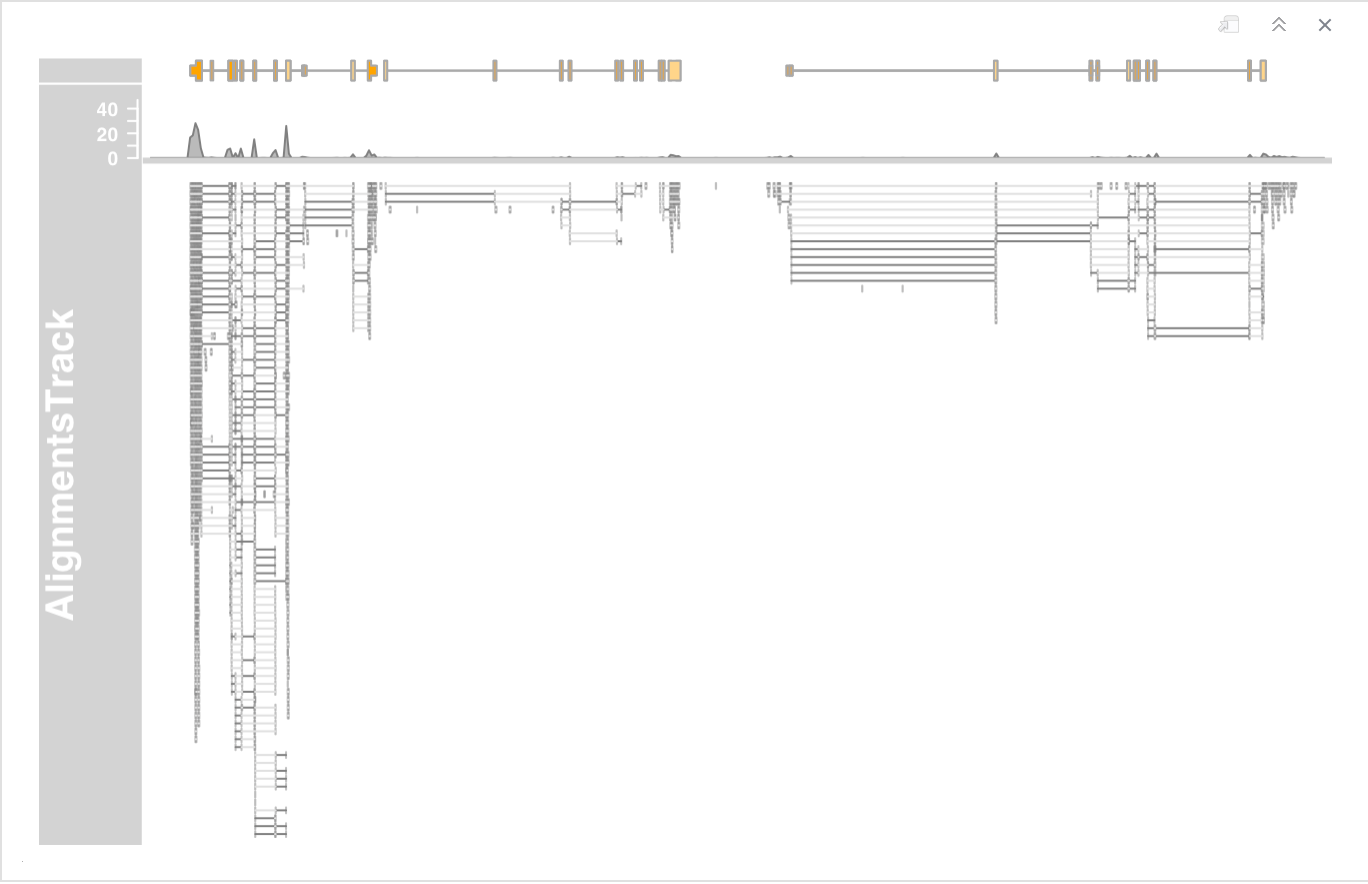
- Turn off the pile-up view of the individual reads by setting the type display parameter.
|
|
output
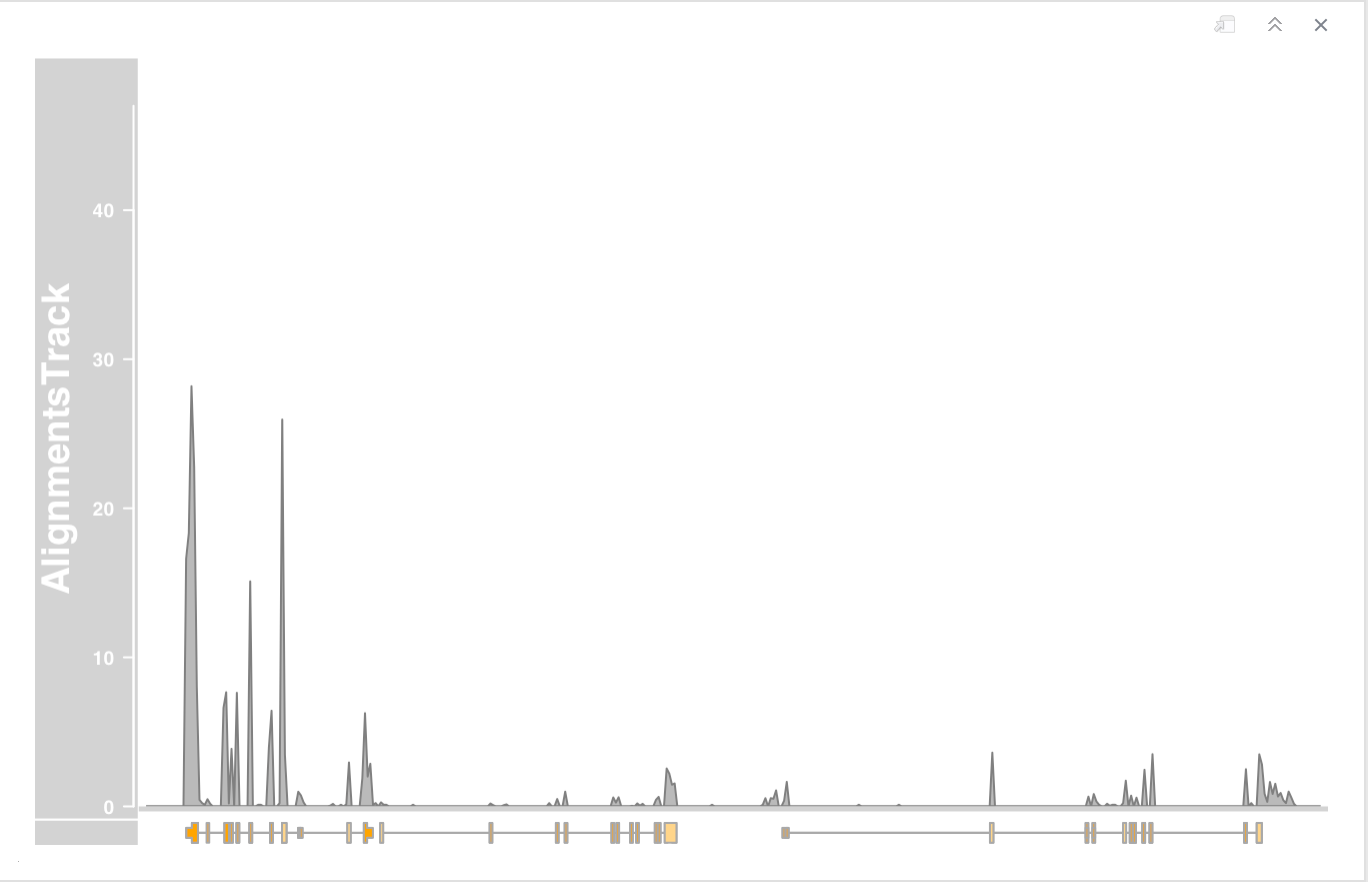
Zoom in a bit further to check out the details of the pile-ups section
|
|
output
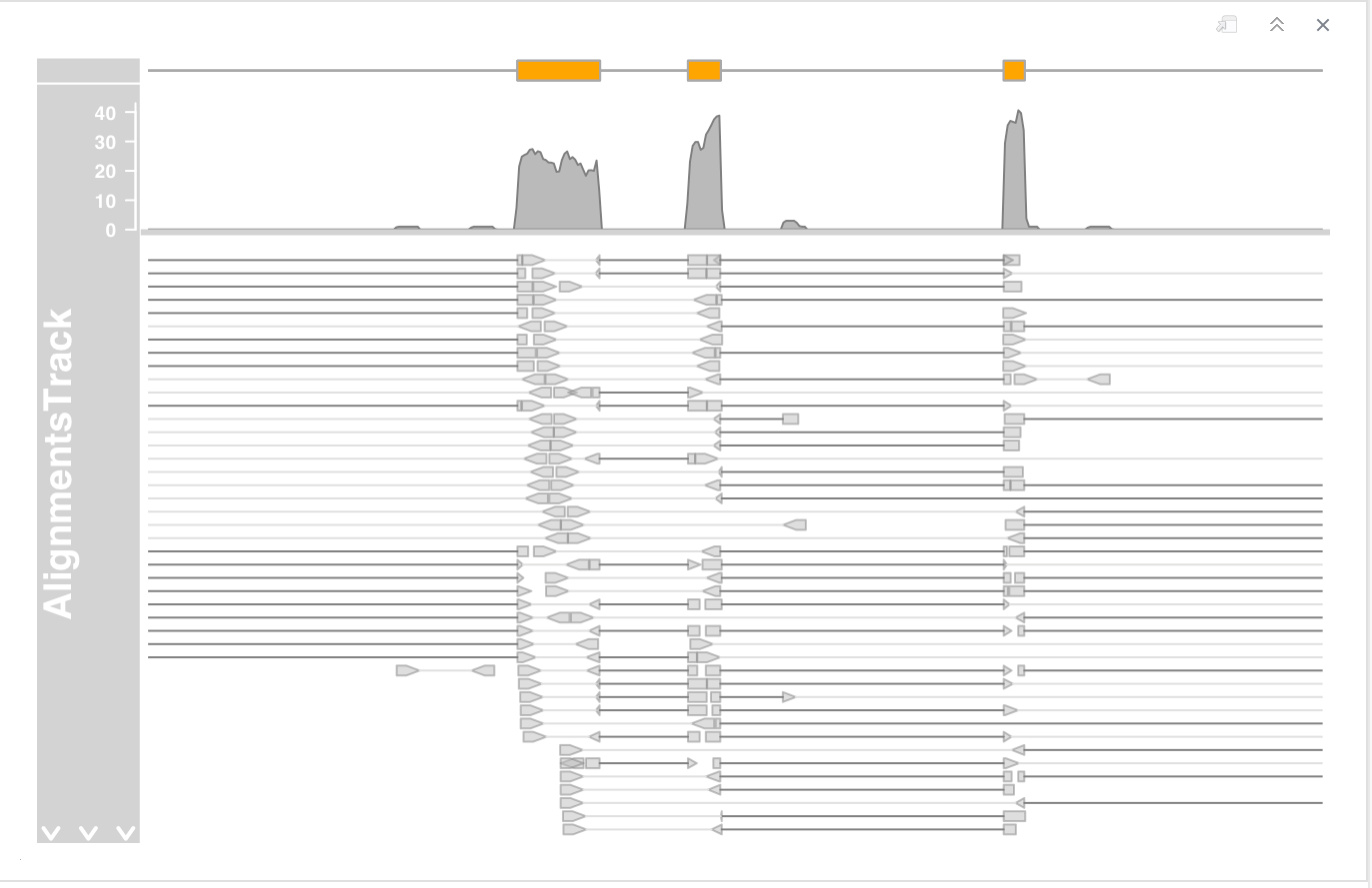
- in single end mode by setting the isPaired argument in the constructor.
|
|
output
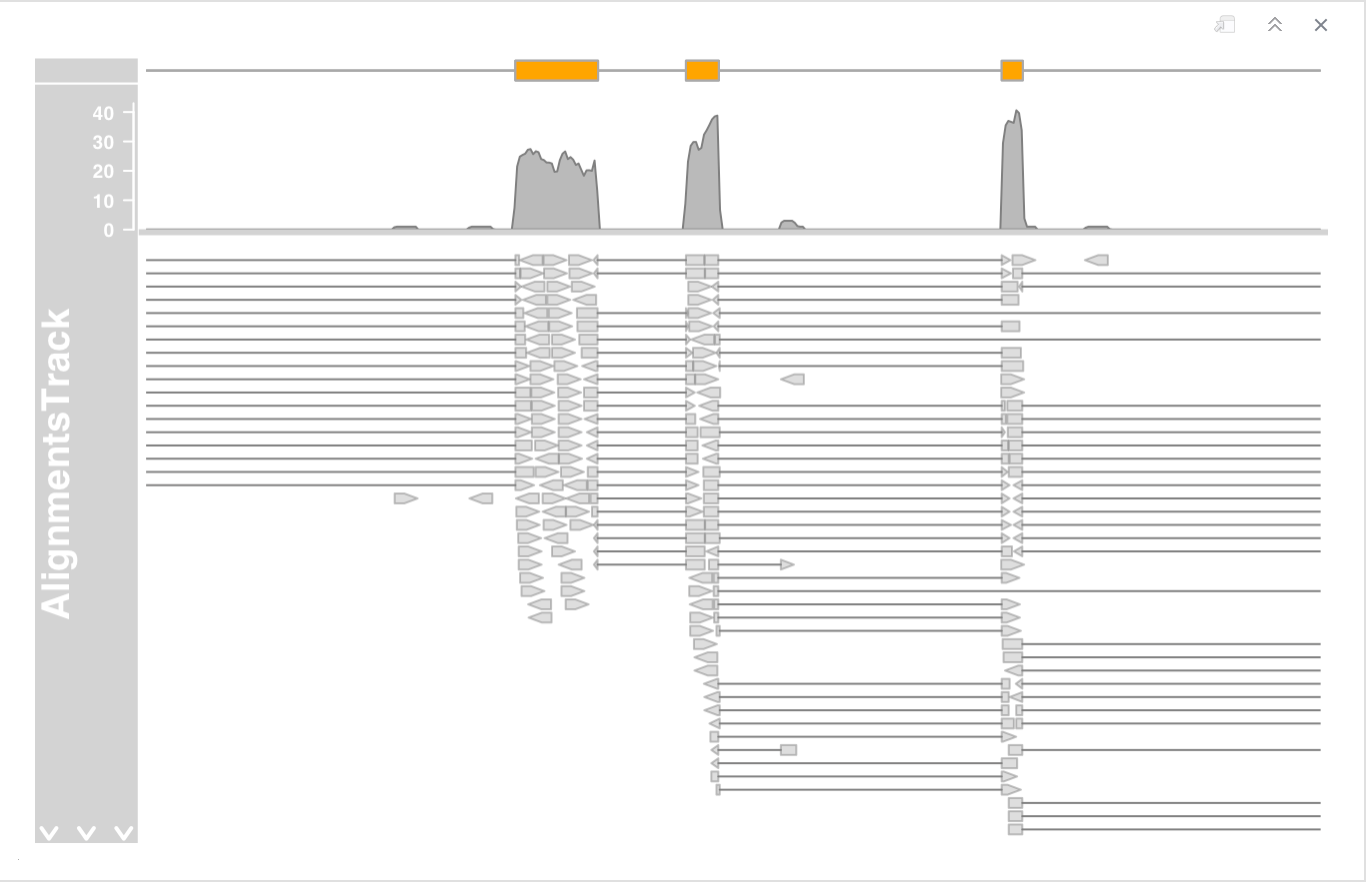
DNAseq experiment
|
|
output
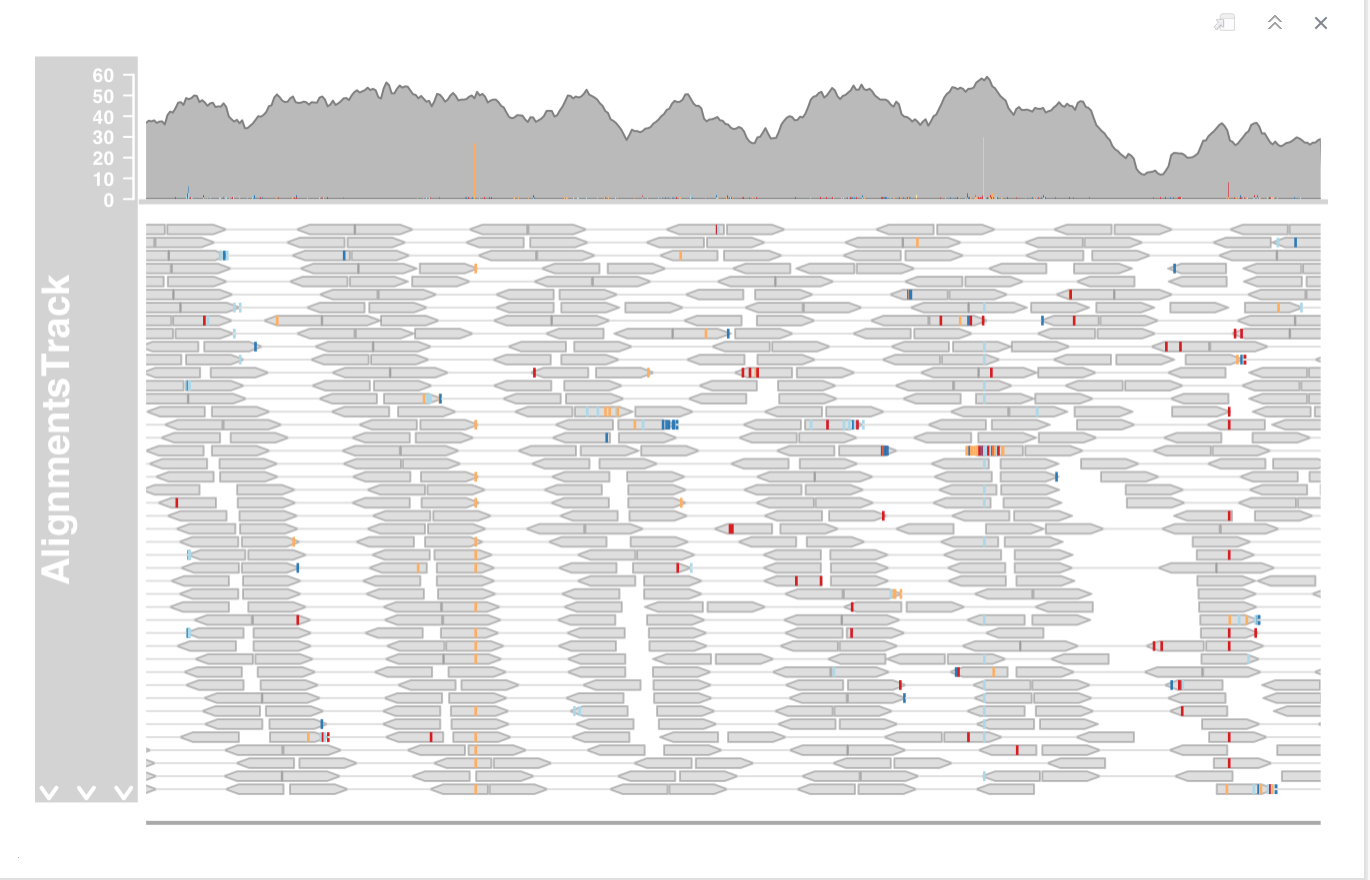
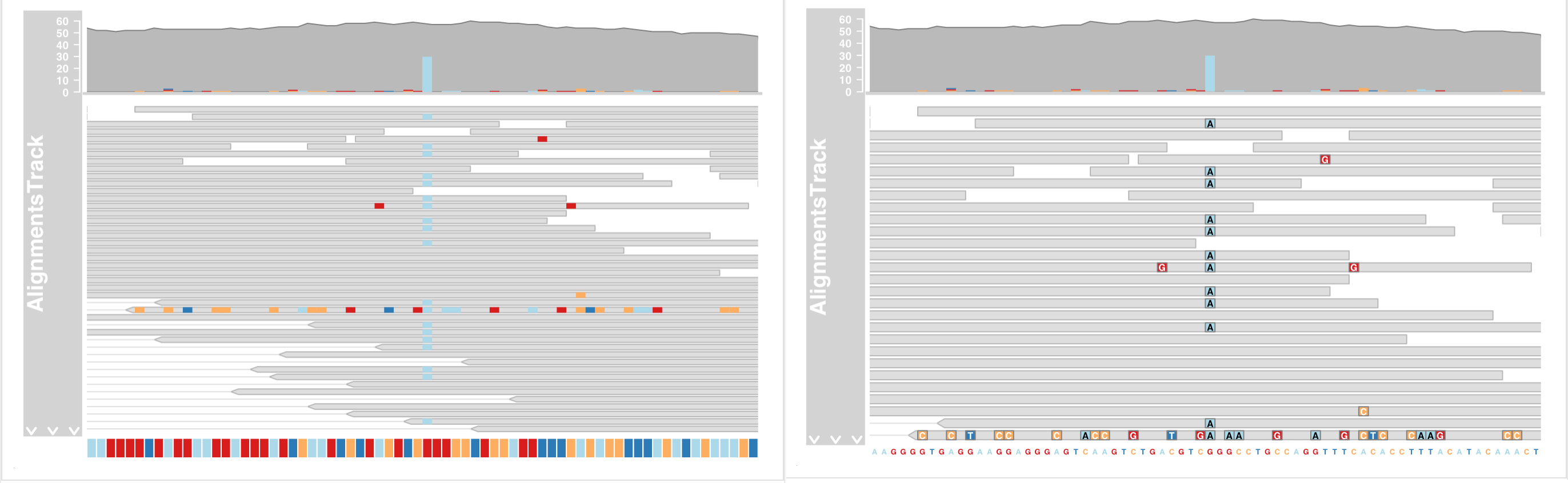
- Zoom in to one of the obvious heterozygous SNP positions
|
|
output
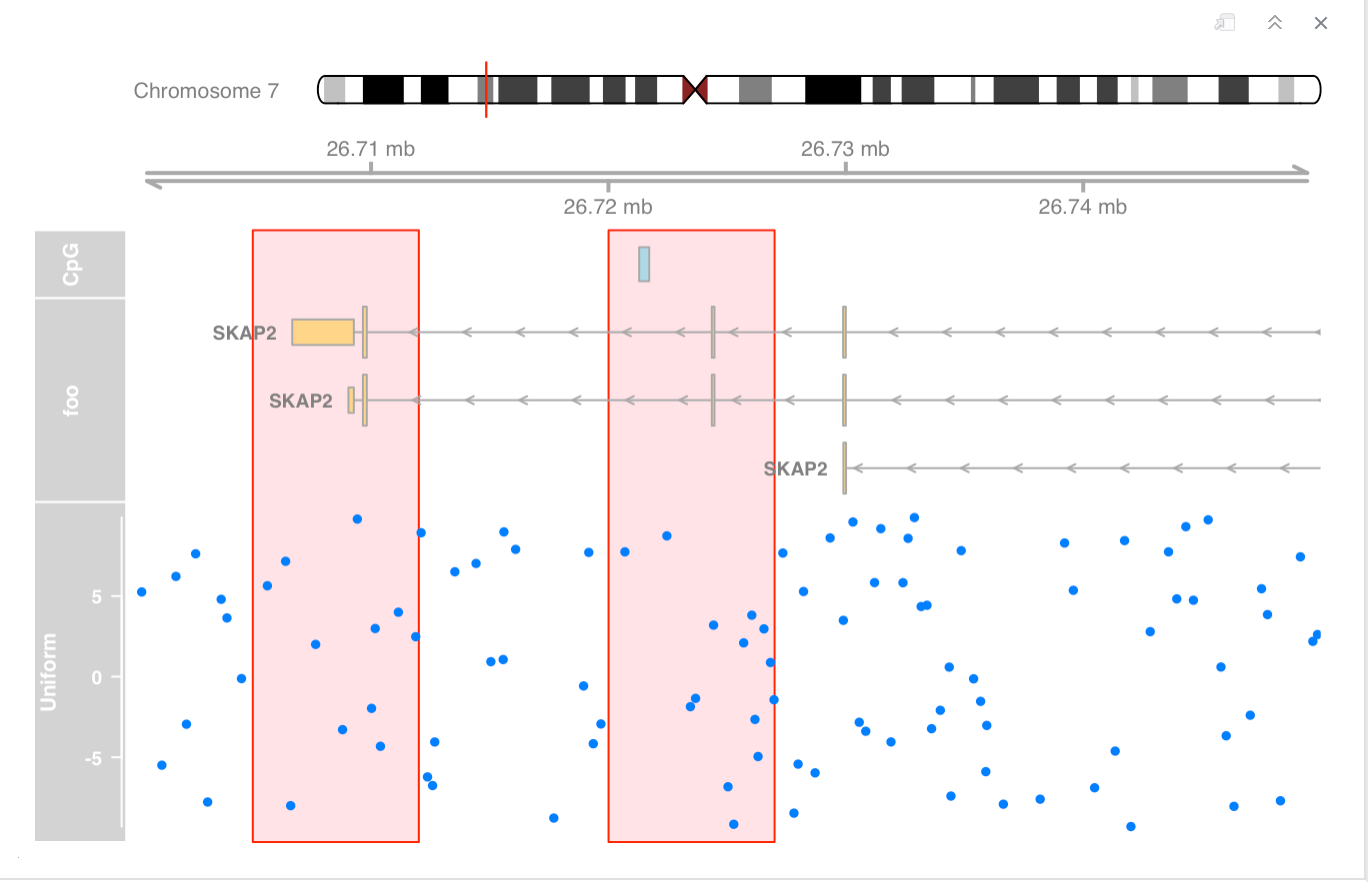
Track highlighting and overlays
|
|
output
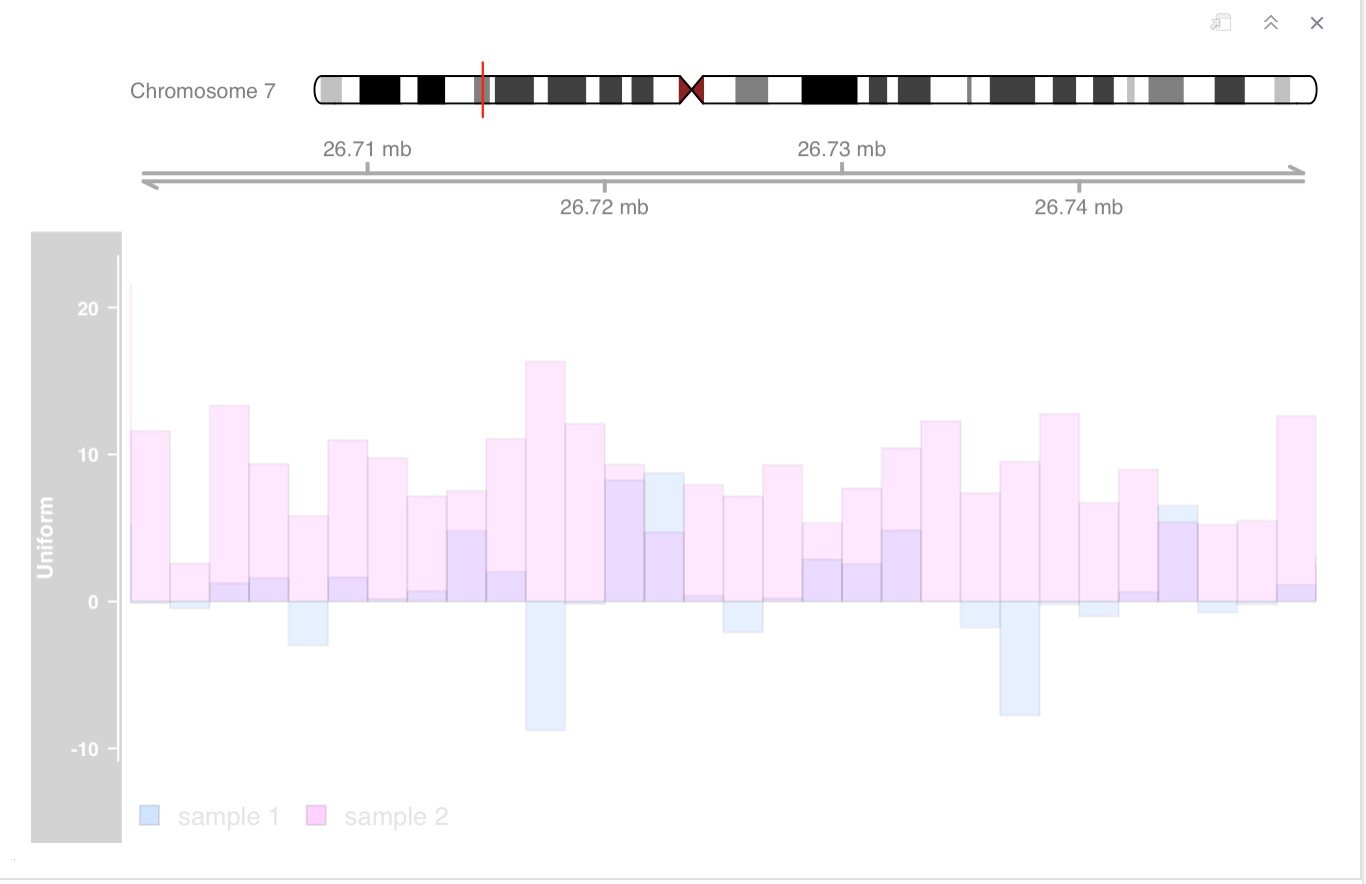
overlays multiple tracks on the same area of the plot
|
|
output
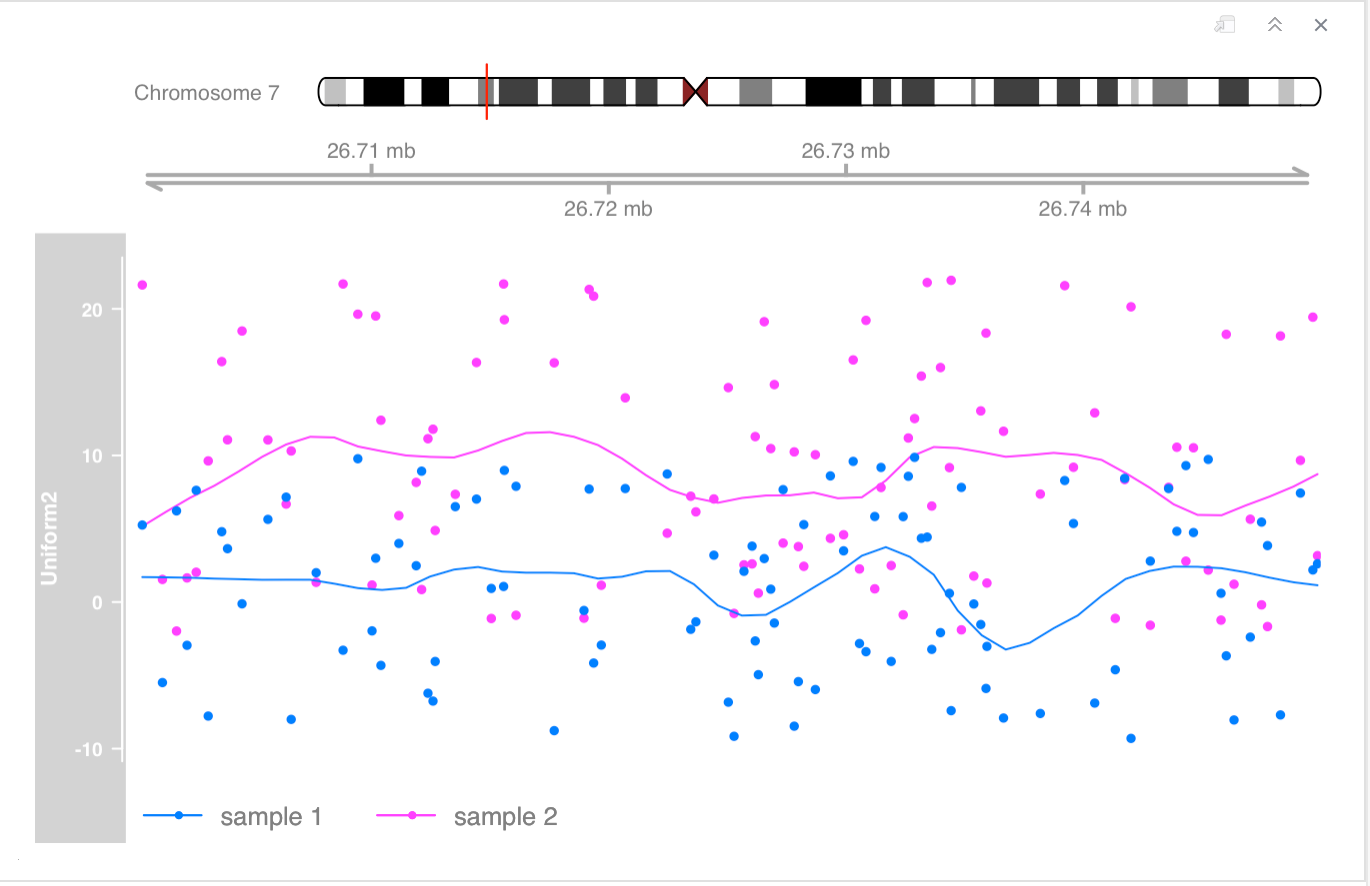
- alpha blending can be a useful tool to tease out even more information out of track overlays
|
|
output
Attach is the .Rmd file.